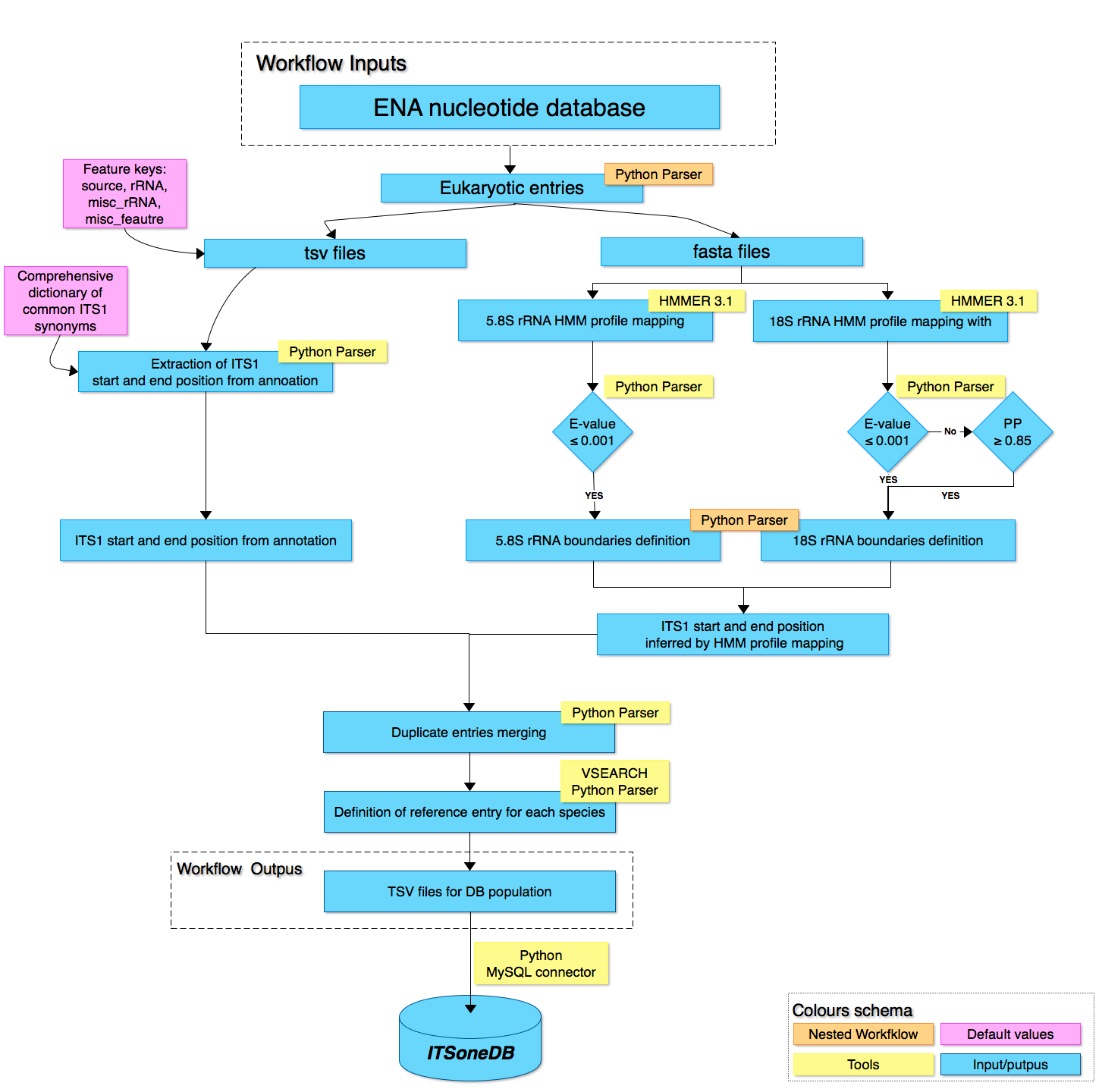
The figure illustrates the main steps deployed in the pipeline procedure for ITSoneDB generation. The software components of the pipeline are implemented in Python programs except the ETL module for database population implemented in Java. In the initial step a collection of Eukaryotis candidate sequences was created by downloading the entire ENA nucleotide release. The collected entries are computed in two different part of the workflow in order to extract or to infer the ITS1 boundaries. On the left side branch, by a pre-compiled dictionary of common ITS1 definition synonymous, the procedure detect and extract ITS1 start and end site from entries feature tables annotations. On the right side branch, ITS1 boundaries are inferred by mapping Hidden Markov Model (HMM) profiles of flanking genes for 18S and 5.8S ribosomal RNA by means of the hmmsearch tool included in HMMER 3.1. The data obtained from the left and the right branch are used to populate the database and entries with both methods informative are merged. Then, a reference entry is defined for each species. At the end, an ETL module combines for each candidate sequence the annotation and the HMM ITS1 boundaries and populates the ITSoneDB by adding further information like species name, taxonomic lineage, ENA description, HMM profile alignments, etc.
Test Case
In order to demonstrate ITSoneDB usability as reference database for ITS1 based metagenomics approaches, we carried out a benchmark test on a real 454 dataset, freely available in the Sequence Read Archive (
SRR174891). The dataset contains 5,160 sequences come from a study of soil of nine T.melanosporum/Q. pubescens truffle-grounds collected in May 2006 in Cahors.
This benchmark test aims shows the effectiveness of ITSoneDB as metagenomics reference database compared to the use of another database, such as UNITE (Kõljalg U. et al, 2005).
The test was carried out by mapping the reads on both ITSoneDB and UNITE databases, using blastn, and considering only significant matches, according to the three criteria:
- at least half of the query sequence must be aligned;
- query and subject sequence must share at least 95% of identity;
- consider significant only subject sequences that are taxonomically informative.
Using ITSoneDB we found 3,206 significant matches while using UNITE we found 2,896 significant matches.
These results demonstrate the ITSoneDB effectiveness as metagenomics reference database for the characterization of fungine populations.
